Introduction to SPATA
SPATA is
a novel hybrid approach that seeks to strike
a good balance between the de novo
and the reference-guided transcriptome
assembly approaches. On one hand, the
difference between SPATA and the de novo
assembly approaches is that SPATA uses the
existing reference genome to divide the
large assembly problem into many smaller
assembly problems and solve them
independently. On the other hand, the
difference between SPATA and the
reference-guided assembly approaches is that
SPATA does not rely on the exact mapping of
the reads to the reference genome.
SPATA is
integrated into assemblySAM. Both GUI
(Mac) and command line (Linux) versions are
available for download.
Hardware & software requirements
System: Mac OS X 64-bit or Linux 64-bit
Recommended Memory: 12GB RAM
Minimum Memory: 8GB RAM
Running SPATA on Linux
1. Install Java 7 on your machine if it is not installed yet. Download assemblySAM-1.1_Linux64.tar.gz for Linux 64-bit. Uncompress the downloaded file by the following command:
tar -zxvf assemblySAM-1.1_Linux64.tar.gz
2. Run assemblySAM.sh using the following command under the assemblySAM folder:
or
java -jar -Xmx8192M assemblySAM.jar -bt2 <bowtie2> -ind <bt2-idx> {-1 <m1> -2 <m2> | -U <r>} - out <out-path> [options]
<bowtie2> bowtie2 executable
<bt2-idx> bowtie2 index filename prefix (without .X.bt2)
<m1> first FASTQ file (paired-end)
<m2> second FASTQ file (paired-end)
<r> single-end FASTQ file
<out-path> output path
Options:
-e <string> error rate (default 0.1)
Examples (paired-end):
java -jar -Xmx8192M assemblySAM.jar -bt2 bowtie2/bowtie2 -ind Examples/bowtieIndex/bowtie2.index -1 Examples/pairedEnd/pairedEnd_1.fastq -2 Examples/pairedEnd/pairedEnd_2.fastq -out Examples/pairedEnd
Examples (single-end):
java -jar -Xmx8192M assemblySAM.jar -bt2 bowtie2/bowtie2 -ind Examples/bowtieIndex/bowtie2.index -U Examples/singleEnd/singleEnd.fastq -out Examples/singleEnd/
Running SPATA on MAC OS
1. Install Java 7 on your machine if it is not installed yet. Download and unpack assemblySAM-1.1_Mac64.zip. Double-clicking on assemblySAM.app application.
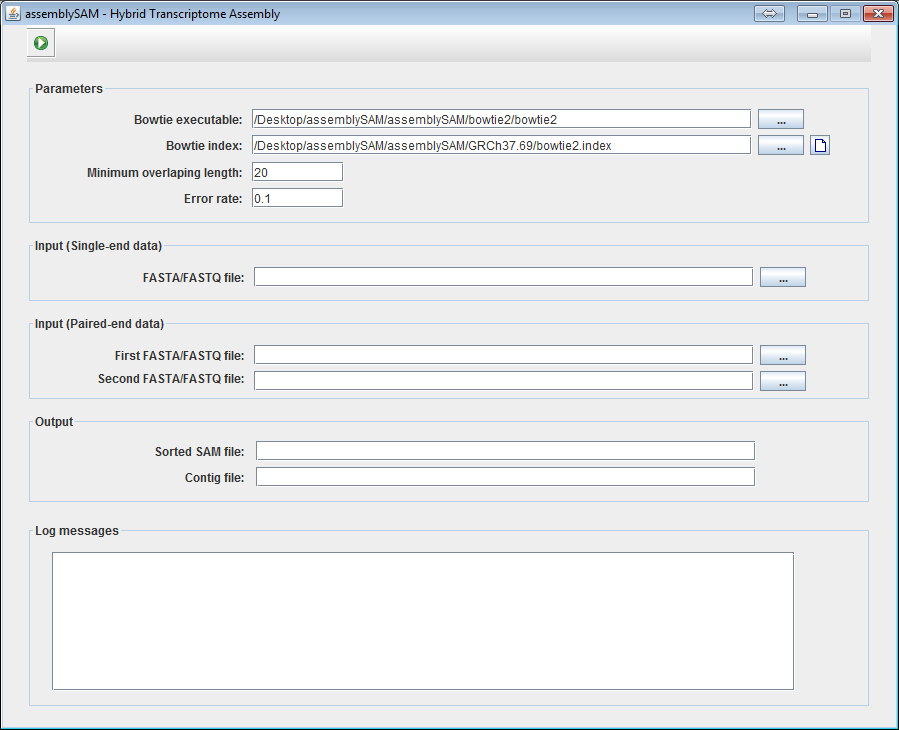
2. Choose the bowtie2 executable location. A bowtie2 executable file is included inside the assemblySAM/bowtie2 folder.
User can also browse and choose an executable file by clicking on the
 button.
button.
3. Choose the bowtie index of the reference genome.

4a. If the User is working with single-end data, give the location of the single-end file.
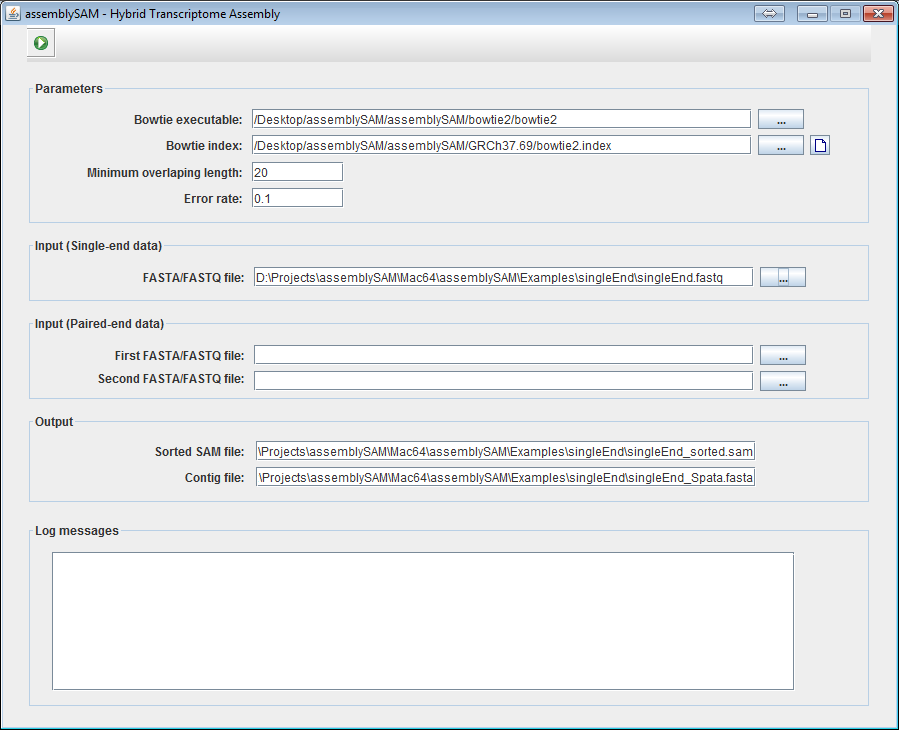
4b. If the User is working with paired-end data, give the location of the two paired-end files.
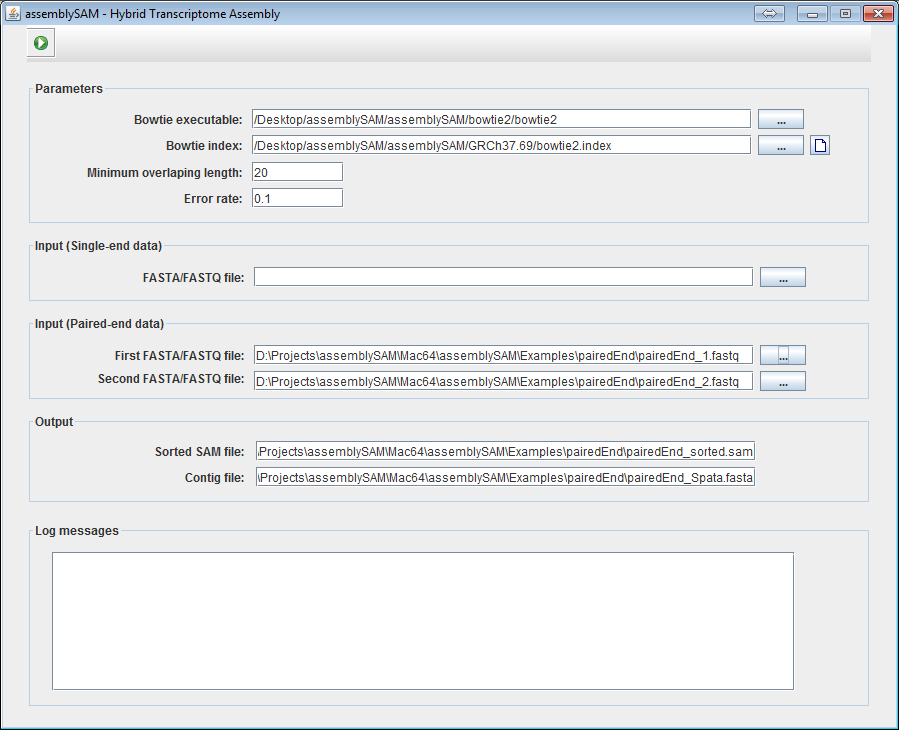
5. Click on "Run" button
 to execute the
procedure.
to execute the
procedure. 6. The localization results and transcript sequences are saved to the files specified in the Output frame.
Inputs of SPATA
Currently SPATA accepts both single-end and paired-end FASTQ file.
Output of SPATA
The software reports 2 outputs:
- Reconstructed transcripts in the fasta
format. The header gives the information
about the chromosome and genomic locus.
- Localization result in the SAM format.