Introduction to SAMMate
SAMMate, a Graphical User Interface (GUI) RNA-seq analysis pipeline, allows biomedical researchers to quickly process fasta/fastq and SAM/BAM files, and is compatible with both single-end and paired-end sequencing technologies. SAMMate implements three mRNA transcript quantification algorithms:
1. SASeq:
A Selective and Adaptive Shrinkage Approach
to Detect and Quantify the active
transcript. Nguyen et al. arXiv:1208.3619
2. RAEM: Read Assignment via EM. Deng et al. Nucleic Acids Research, doi:10.1093/nar/gkr042.
Manual:
SAMMate manual
Screenshot:
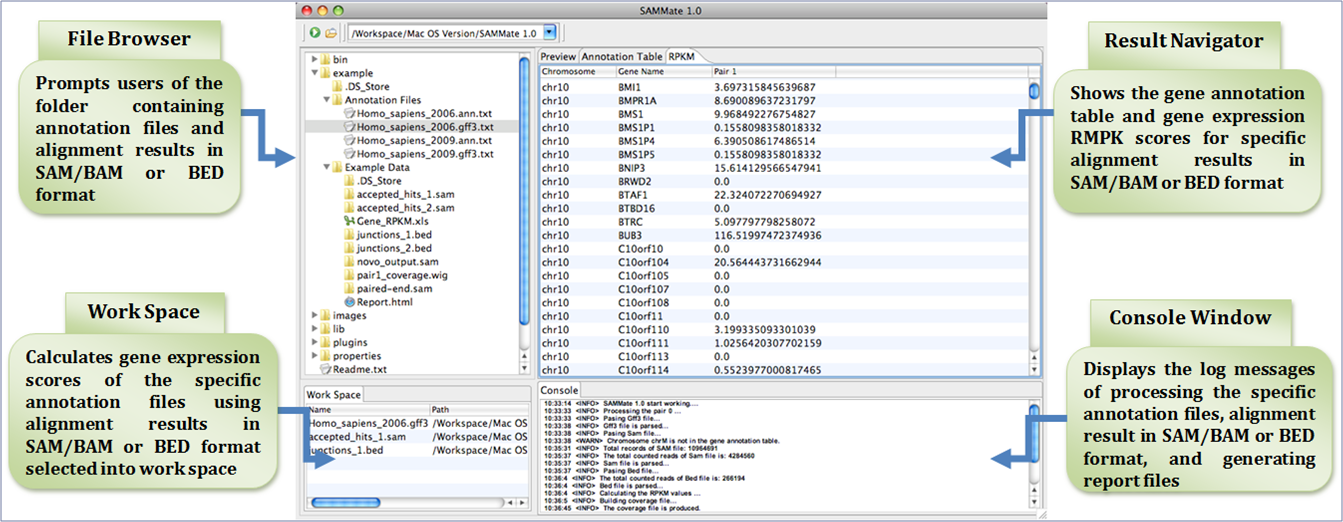
Feature list:
- Estimating genomic feature abundance at isoform level
- Calculating genomic feature abundance score at gene level
- Generating signal map for peak detection
- Generating wiggle files for visualization
- Supported Fastq or Fasta format by calling Bowtie to align short reads to a reference genome
- Generating alignment report
- Customize gene annotation file
- Customize chromosome name in output file
- SAM/BAM format conversion
- SAM/BAM file sorting
- Supports edgeR to detect differentially expressed genes and isoforms.
Update news:
Version 2.7
- Supports edgeR to detect differentially expressed genes and isoforms.
- Supported simultaneous multiple instances of SAMMate runs.
- Users can export P-values to transcript expression file.
- In the Work Space, added two group tabs to separate files into two groups.
- In the Options dialogue, added R Options to configure R information
Frequently
asked questions:
How many
formats of gene annotation file can be
supported by SAMMate?
SAMMate can support 4 gene annotation files in
GTF, GFF3, refFlat and GeneBank format. Please
use .ann as the extension of refFlat format
and use .seq as the extension of GeneBank
format.
Can SAMMate
handle paired-end alignment results in the
SAM/BAM format?
Yes, SAMMate can automatically detect if the
inputting SAM/BAM files are paired-end or
single-end.
Can SAMMate
start with Fasta/Fastq file?
Sure. Since from version 2.6, SAMMate has
integrated the Bowtie aligner to process
Fasta/Fastq sequence files. For large
Fasta/Fastq sequence files, the Mac and Linux
version of SAMMate are recommended for
efficiency and stability of the Bowtie
aligner.
Can SAMMate
process a batch of SAM/BAM files ?
Yes, SAMMate can process a batch of SAM/BAM
files selected in "Work Space".
Can SAMMate
process a pair of SAM/BAM and BED files ?
Yes, SAMMate can process the combination of
SAM/BAM and BED file. When users input the
SAM/BAM and BED files into the "Work Space",
SAMMate can automatically process the SAM/BAM
and BED files by the order of files, and the
first SAM/BAM file and the first BED file
would be the first combination, and the second
SAM/BAM file plus the second BED file would be
the second combination, and so on.
Can SAMMate
support simultaneous multiple instances of
SAMMate runs?
Yes. SAMMate 2.6.1 support this feature.
How do I find
differentially expressed genes and
transcripts with count-based?
You need to enable using edgeR in the Options
dialogue, and then assign your sequence files
into two different groups. SAMMate would
automatically export the differential
expressed genes and transcripts files using
the counts of short reads that mapped in the
genes and transcripts.
Where can I
find the resulting files?
The resulting files reported by SAMMate would
be generated in the directory of the inputted
gene annotation file.